README for rSeq: RNA-Seq Analyzer
rSeq is a set of tools for RNA-Seq data analysis. It consists of programs that deal with many aspects of RNA-Seq data analysis, such as reference sequence generation, sequence mapping, gene and isoform expressions (RPKMs) computation, etc. There are also many other features that will be gradually added into rSeq in the future.
Simply download the zipped file from the rSeq website and unzip the file to somewhere on the hard drive. If you downloaded the source code, simply type "make" to compile it. You will need a recent C++ compiler which supports C++11 standard.
Gene and isoform expression estimation
The procedure for gene and isoform expression estimation consists of three steps: transcript sequence preparation, read mapping and expression estimation.
Note: since rSeq 0.1.0 a new way for generating transcripts and estimating gene and isoform expression values is used. The old way (here are the old user manual and old FAQ) is still supported but not recommended.
[1] Prepare transcript sequences.
Transcript sequences should be stored in a file in the FASTA format. Name different transcripts (isoforms) of a gene as gene_name$$isoform_id such as WASH5P$$NR_024540 and here is an example. Note: gene_name will be the index used by rSeq to output gene expression. So if you want something other than gene names, such as Entrez gene ID, you should use them in the transcript sequence file.
If you can prepare transcripts according to the above format then you can jump to step 2 directly. rSeq also provides two methods for transcript sequence generation. The first method is easier but it only works with the UCSC refFlat annotation. The second method is slightly more complicated but it is recommended, and it works with gene annotation in the GTF format (e.g., the ENCODE annotation), UCSC refFlat format and UCSC KnownGene format (BED format). The second method is also required to generated alignment results in BED files (described in more details in a later section).
rseq annotate_transcripts refMrna.fa kgXref.txt
Where "refMrna.fa" is the unzipped file containing transcript sequence downloaded from the UCSC genome browser. "kgXref.txt" is the unziped file containing the gene name/transcript id corresponding table also download from the UCSC genome browser. For human genome, it is at http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/kgXref.txt.gz. When finished, you will get a "refMrna.fa.new.fa" file, which contains all the annotated transcript sequences in FASTA format.
rseq
generate_transcripts
refFlat.txt chrfilelist.txt
where "refFlat.txt" is the annotation file in UCSC refFlat format and "chrfilelist.txt" is the list of filenames of DNA sequences of all the chromosomes. And here is an example. When finished, you will get a "refFlat.txt.fa" file, which contains all the transcript sequences in FASTA format.
Note: starting from rSeq 0.2.1, generate_transcripts also accepts the whole genome in a single fasta file, i.e.
rseq generate_transcripts refFlat.txt genome.fasta
From now on I will demonstrate using rSeq with transcript sequences generated with method 1. If you are using method 2, simply replace all the "refMrna.fa.new.fa" with "refFlat.txt.fa" in the command lines.
[2] Map the reads to the transcript sequences.
The rSeq package includes SeqMap as the default aligner, which usually works well with ~10M reads for desktop PCs with 2GB memory. For larger data sets, you can either use a computer with larger memory or split the read file into several files, map each file respectively and then concatenate the final mapping results as one single file.
If you can handle it technically, it is recommended that you use other aligners which output in either the SAM format or the Eland-multi format. We have tested rSeq with SeqMap, Eland, BWA, Bowtie and Bowtie2. Aligners other than SeqMap need to be installed by yourself. Simply go to the aligner's website and follow its instructions to download and install it. Each aligner has its own options and features. Here I only summarize the most basic usage of a few aligners. For better control of the alignment procedure or advanced features, please follow the user manual of the particular aligner that you are using.
seqmap 2 reads.input refMrna.fa.new.fa reads.mapped.output /eland:3
Where "reads.input" is the file containing the sequencing reads (in plain read sequences, FASTA or FASTQ format, see SeqMap's user manual for more details). refMrna.fa.new.fa" is the annotated transcript sequence file. "reads.mapped.output" is the output file. For paired-end reads, map the two files for each end separately. Please refer to SeqMap's user manual for more options.
bwa index refMrna.fa.new.fa
Then use the following
commands to align single-end reads:
bwa aln refMrna.fa.new.fa reads.fastq >
reads.bwa.sai
bwa samse -n 100 -f reads.mapped.sam refMrna.fa.new.fa reads.bwa.sai reads.fastq
Where "reads.fastq"
is the read file in the FASTQ format.
BWA (and Bowtie and Bowtie2) also
accept FASTQ or other input formats. See their user manuals
for the usages. "reads.mapped.sam"
is the output file in SAM format. For paired-end reads, either map
files for each end separately, or use the following commands:
bwa aln refMrna.fa.new.fa reads_1.fastq >
reads_1.bwa.sai
bwa aln refMrna.fa.new.fa reads_2.fastq
> reads_2.bwa.sai
bwa sampe -a 10000 -n 100 -f reads.mapped.sam refMrna.fa.new.fa reads_1.bwa.sai
reads_2.bwa.sai reads_1.fastq reads_2.fastq
Please refer to BWA's user manual for more options.
bowtie-build refMrna.fa.new.fa refMrna.fa.new.fa
Then use the following command to align single-end reads:
bowtie -k 100 -S refMrna.fa.new.fa reads.fastq reads.mapped.sam
For paired-end reads, either map files for each end separately, or use the following command:
bowtie -k 100 -S refMrna.fa.new.fa -1 reads_1.fastq -2
reads_2.fastq reads.mapped.sam
Please refer to Bowtie's
user manual for more options.
bowtie2-build refMrna.fa.new.fa refMrna.fa.new.fa
Then use the following command to align single-end reads:
bowtie2 -x refMrna.fa.new.fa -U reads.fastq -S reads.mapped.sam
For paired-end reads, either map files for each end separately, or use the following command:
bowtie2 -x refMrna.fa.new.fa -1 reads_1.fastq -2
reads_2.fastq -S reads.mapped.sam
Please refer to Bowtie2's
user manual for more options.
eland reads.txt refMrna.fa.new.fa.squashed/ reads.mapped.output --multi
Where "reads.txt" is the file of the sequence reads in plain read sequences. refMrna.fa.new.fa.squahshed/" is the directory containing the squashed sequences generated with the "squashGenome" tool in the Eland package. For paired-end reads, either map files for each end separately.
[3] Compute gene and isoform expressions.
Use the following command:
rseq expression_analysis refMrna.fa.new.fa mapped.reads.output1 [mapped.reads.output2]
where "mapped.reads.output1" (and also mapped.reads.output2 for paired-end reads when each ends are mapped individually) are the output files from the aligners, in either the Eland-multi format or the SAM format. When finished, you will get an output file: "mapped.reads.output1.exp.xls" (together with several other files for diagnostic purpose which you should simply ignore). It contains the estimated gene and isoform expression values, indexed by gene names and isoform IDs. It is a tab-delimited text file with one gene per line and can be opened by any text editor or Excel. For example, the following line means that gene SDF4 has an expression estimate 103.293, in the unit of RPKM. The gene has two isoforms NM_016547 and NM_016176 with expression estimates 0 and 103.293 respectively.
SDF4 103.293 2 NM_016547,NM_016176 0,103.293
Here is an example of the output file.
[4] Generate BED alignment file.
rSeq can generate alignment file in UCSC BED format, which can be visualized in UCSC Genome Browser as a custom track or in CisGenome Browser. Use the following command:
rseq expression_analysis -a refFlat.txt refFlat.txt.fa mapped.reads.output1 [mapped.reads.output2]
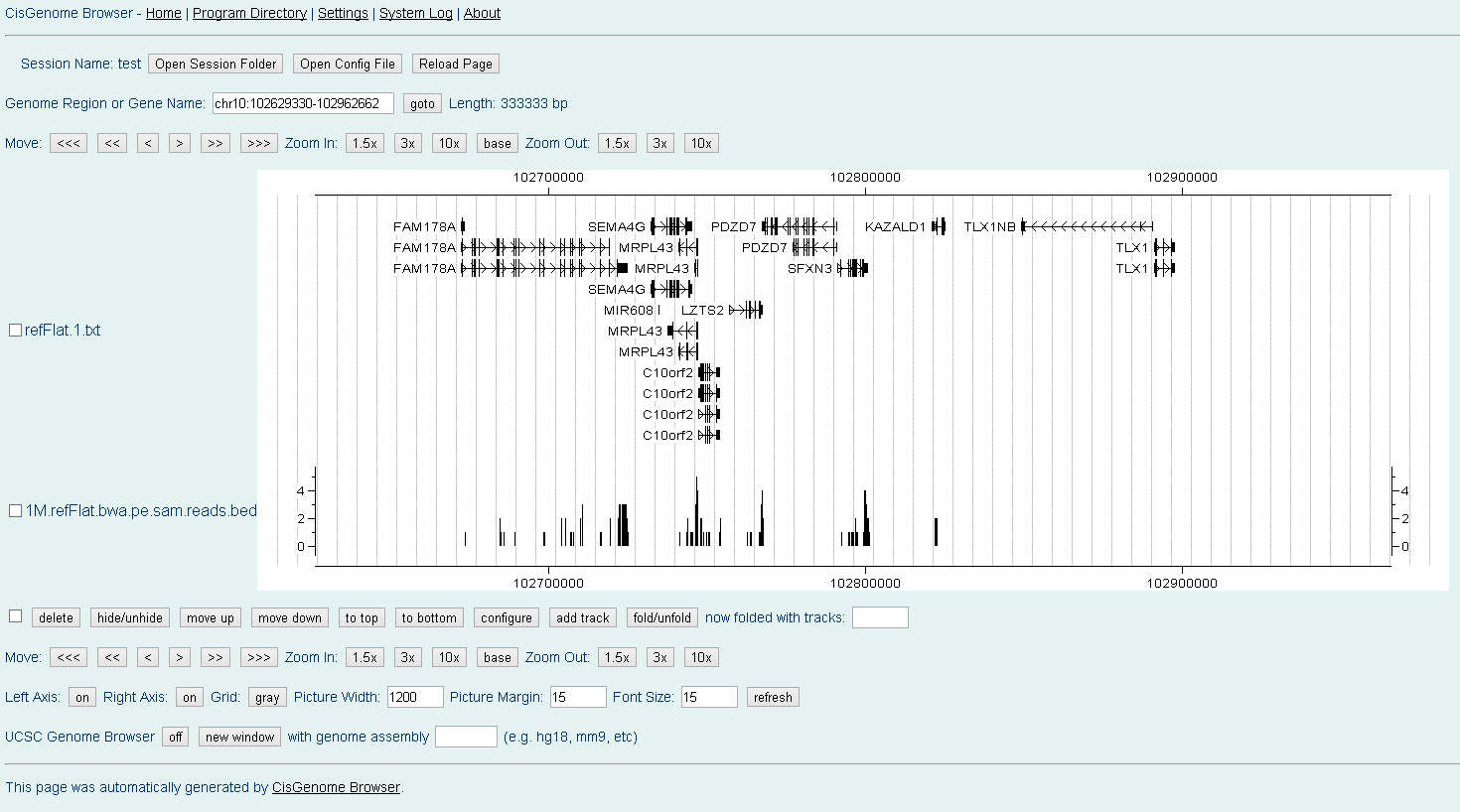
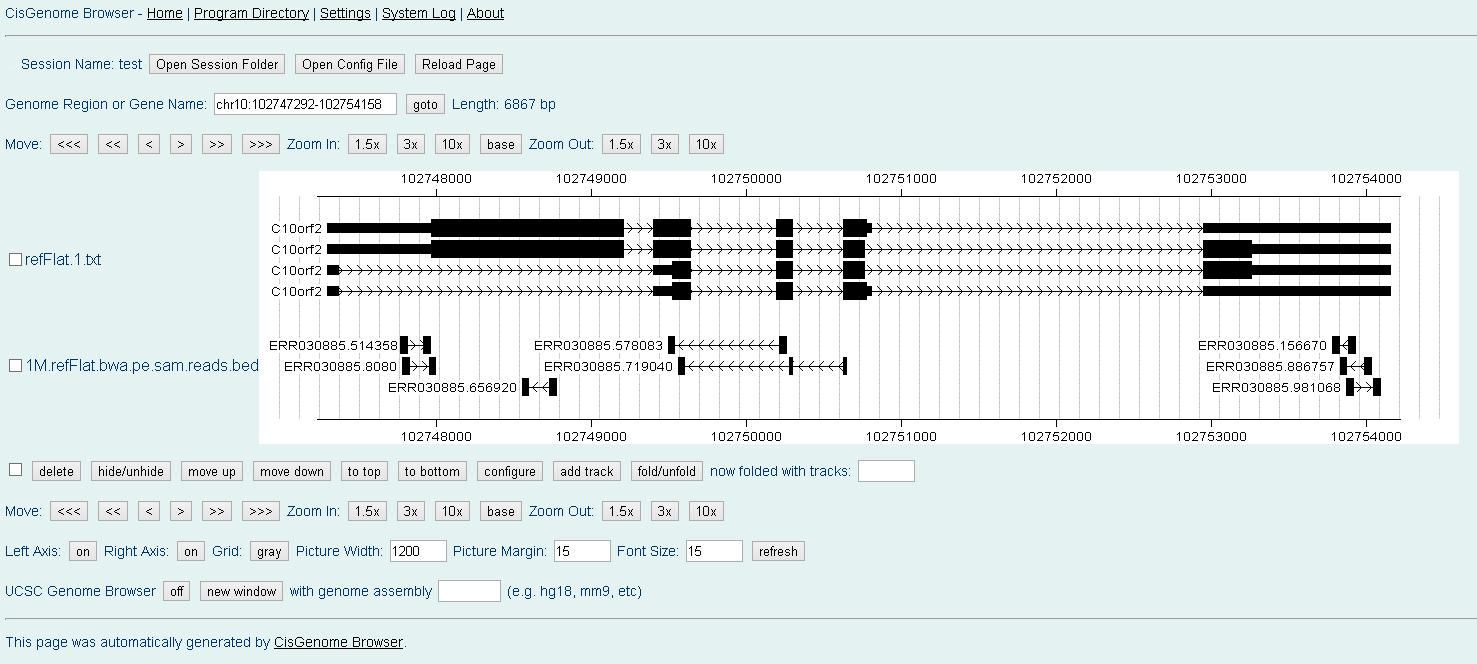
where "refFlat.txt" is the gene annotation file in UCSC refFlat format and "refFlat.txt.fa" is the transcript sequence file generated using with the second method, i.e., "rseq generate_transcripts refFlat.txt chrfilelist.txt". When finished, you will get a "mapped.reads.output1.reads.bed" file, which contains all the uniquely aligned reads in UCSC BED Format. Here and Here are two schreenshots of visualizing aligned paired-end RNA-Seq reads in CisGenome Browser.
Anyone can use the source codes, documents or the excutable file of rSeq free of charge for non-commercial use. For commercial use, please contact the author.
For isoform-specific gene expression estimation, rSeq uses the method described in
Jiang, H., Wong, W.H. (2009) Statistical Inferences for Isoform Expression in RNA-Seq, Bioinformatics, 25(8), 1026–1032. [online]
For paired-end RNA-Seq analysis, rSeq uses the method described in
Salzman, J., Jiang, H.,
Wong, W. H. (2011) Statistical
Modeling of RNA-Seq Data, Statistical Science, 26
(1): 62-83. [online][arxiv]
rSeq was developed and tested with the help of the members and several collaborators of the Wong lab.
{kind=link}
{kind=link}